Decentralised clinical trials (DCTs) are not new, though their popularity and utilisation has grown since the COVID-19 pandemic. DCTs are enabled by and mutually reinforce an expanding innovative “digiverse” that includes all digital technologies that can be customised to support biomedicine and clinical research. This digiverse is changing and expanding rapidly, and regulators have been working to maximise the benefits. The use of digital health technologies (DHTs) has implications for a number of key areas, and regulators have already provided guidance for diversity and inclusion in clinical trials, privacy considerations and endpoint selection, among others.
Decentralised clinical trials technology ecosystem at a glance
The decentralised clinical trials industry is projected to have a compound annual growth rate (CAGR) of between 6.7 and 14.8% until 2027.[1],[2] The various digital technologies that support DCTs are projected to experience a staggering CAGR of 26.9% from 2022 to 2030.[3]
Figure 1 illustrates the DCT ecosystem, which, to ensure smooth operations and the proper flow of information, should be customisable, modular, integrated and interoperable. It must also comply with the US Code of Federal Regulations, Title 21, Part 11, Good Clinical Practice (GCP), and the privacy principles of the General Data Protection Regulation (GDPR) in the EU and the Health Insurance Portability and Accountability Act of 1996 (HIPAA) in the US.
The system design should provide a single interface for trial participants and, of course, be easy for investigators and site staff to use.
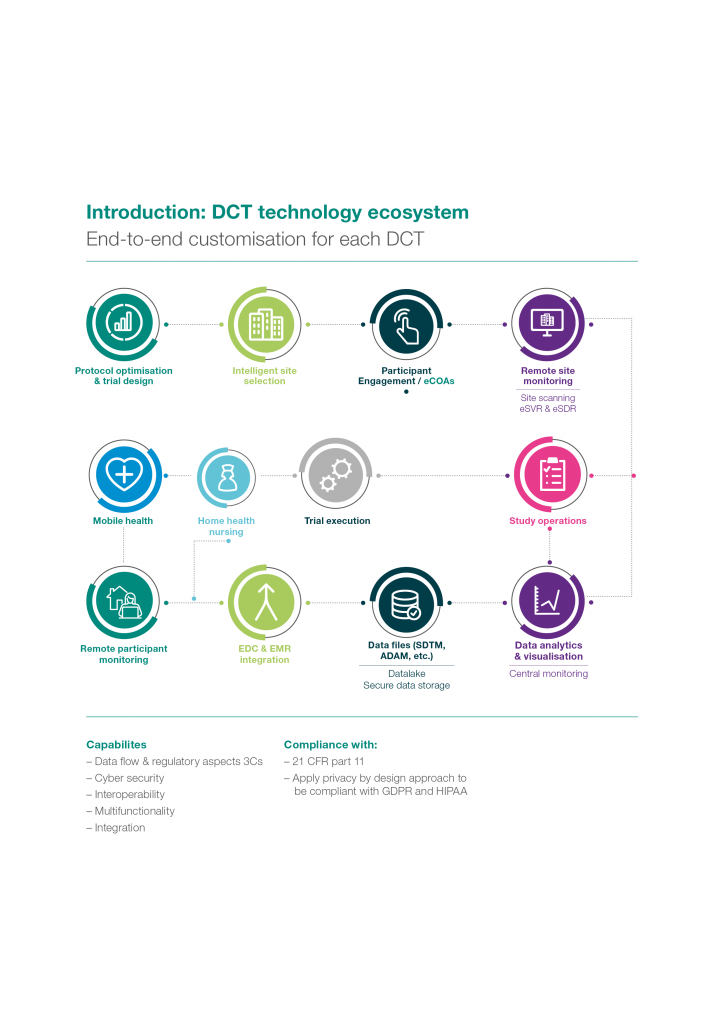
Figure 1 | The DCT technology ecosystem
Regulators’ commitment to DHT
The United States Food and Drug Administration (US FDA) published draft guidance on the use of DHTs in traditional and decentralised trials in May 2023, with final guidance expected not later than December 29, 2023.[4] Topics in the draft guidance for DCT include:
- Design
- Conduct of remote clinical trial visits and clinical trial-related activities
- Use of DHTs to remotely acquire data
- Roles and responsibilities of the sponsor and investigators
- Obtaining informed consent and institutional review board oversight of the informed consent process
- Determination of the appropriateness of investigational products for use
- Packaging and shipping of investigational products
- Safety monitoring of trial participants
- Software used to support the conduct
The FDA has created the Digital Health Centre of Excellence (within the Centre for Devices and Radiologic Health) to support the use of decentralised clinical trials in product approvals as well as to maintain an inventory of related guidance.
The European Medicines Agency (EMA) has published a guidance in 2023 and Q&A document on qualifying novel methodologies. This outlines a pre-approval procedure for the use of a variety of digital tools via an EMA/Committee for Medicinal Products for Human Use (CHMP) qualification opinion. This is similar to the scientific advice procedure in the EU and the Innovative Science and Technology Approaches for New Drugs (STAND) program managed by the FDA. Because gaining pre-approval is a protracted process, sponsors should fully understand what is entailed before proceeding.
A path to equity, inclusion and diversity
One of the technology-enabled value propositions of decentralised clinical trials is enhanced equity, diversity and inclusion within the trial population. This benefit is only realised, however, if the “digital health divide” is considered and all barriers to participation are mitigated, including access to and use of these technologies.
The FDA Reauthorisation Act (FDARA) encourages sponsors to draft development plans that include diversity in clinical trials. In 2022, the FDA issued a draft guidance to industry[5] for developing plans to enrol more participants from underrepresented racial and ethnic populations into clinical trials.
Facilitating consent
Technology can facilitate the informed consent process by providing engaging, accessible information to potential participants, allowing consent to be granted anywhere, and archiving multiple versions of consent forms over the lifecycle of the trial.
The relevant regulations (Figure 2) cover eSignatures, data privacy, retention of materials and the qualification and validation of technology components of any eConsent solution. Regulations are often country specific, and thus it is important to have country-level expertise involved from the design of the solution through to the end of the trial. Belgium, Denmark, Israel and Sweden have requirements or guidelines specific to eConsent, and it is expected that the EMA will provide more guidance in the future.
The FDA published a comprehensive Q&A document on eConsent in 2016 (Use of Electronic Informed Consent Questions and Answers), which is recommended reading for anyone contemplating using eConsent.
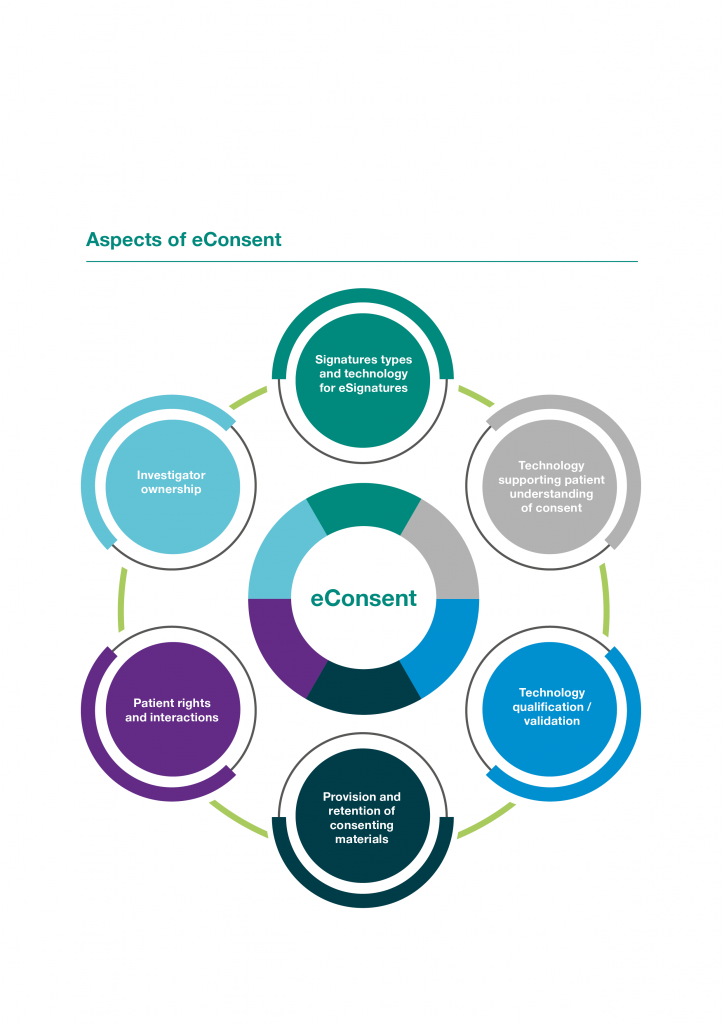
Figure 2 | Aspects of eConsent
Using a bring-your-own-device (BYOD) approach
Clinical data can now be collected via electronic outcomes assessments (eCOA) via patients’ own mobile devices or computers. The EMA has released final guidance (EMA/INS/GCP/112288/2023) on computerised systems and electronic data in clinical trials. The recent guidance by EMA and FDA are being considered by other countries as the foundation for their requirements.
Sponsors should carefully consider the capabilities of any BYOD technology and confirm that it can support the needed endpoint. This begins with defining the DHT endpoint and then validating it against any existing alternative endpoint. A novel endpoint must be justified in the statistical analysis plan (SAP), showing that it is a statistically meaningful measure of severity or status, explaining how it relates to other effective endpoints for similar conditions, and testing it against a similar endpoint in a licensed drug (if possible).
Practical considerations for using DHTs
Medical grade vs. health/wellness devices
While most sponsors would likely prefer to use consumer grade health and wellness devices as the requirements are less demanding, a device falls into the medical grade category if the manufacturer is making medical claims for it, if it fits within the standard definition of a medical device and if it will aid in clinical decision-making. General wellness devices, however, are those that encourage a healthy lifestyle and are not related to diagnosing, curing, mitigating, preventing or treating a disease or condition.
CE marking
The European Commission CE mark is only obligatory for products for which EU specifications exist and require the affixing of CE marking. When CE marking is required, the product must follow all applicable EU requirements.
Fit-for-purpose
Sponsors must ensure that devices can be used safely and effectively by the user population in the intended environment, and include the information needed to use them safely and properly.
Risk assessment
Once the device’s use is determined and the patient population and use environment are defined, sponsors must perform a safety and security risk assessment. Sponsors must develop a mitigation plan based on the risk assessment.
Data privacy
Given the increasingly complex global privacy legal landscape, the associated scrutiny of large corporations from data protection supervisory authorities, and the risk of large fines for non-compliance, ensuring privacy-by-design principles are embedded in the development and rollout of DCTs is crucial.
The GDPR is widely recognised as one of the most robust regulations for personal data protection. Thus, a rule of thumb is that compliance with GDPR will ensure compliance with every other privacy law.
Conclusion
The DHTs that make decentralised trials possible must be fit for purpose and comply with regulations across all jurisdictions in the trial footprint. Therefore, having in-country regulatory intelligence is critical to successfully applying technology in DCTs. The goal of the expanding array of DHT is to facilitate compliance with study protocol. Prioritising and demonstrating privacy compliance in the design and rollout of DCTs will increase and support wider usage.
This article was authored by Jo Hulbert, Executive Director, Global Regulatory Clinical Services with contributions from Arwa Shurrab, Senior Director, Global Regulatory Affairs and Priti Prasad, Senior Manager, Global Regulatory Affairs, all at ICON.
References
[1] Decentralized Clinical Trials (DCTs) Market 2022-2028: Share, Trend, Industry News, Demand, Business Growth, Top Key Players Update, Business Statistics and Research Methodology. Globe Newswire, April 2022
[2] Virtual Clinical Trials Market Size to Reach US$ 21.5 Bn by 2030. Biospace, May 2022
[3] U.S. Digital Health Market Size, Share & Trends Analysis Report by Technology, by Component and Segment Forecasts, 2022 – 2030. Globe Newswire, March 2022
[4] FDA Draft Guidance for Industry, Investigators, and Other Stakeholders Decentralized Clinical Trials for Drugs, Biological Products, and Devices, May 2023
[5] FDA Draft Guidance for Industry: Diversity Plans to Improve Enrollment of Participants from Underrepresented Racial and Ethnic Populations in Clinical Trials, April 2022